Understanding the Basics of Amino Acids in Protein Chemistry
When it comes to molecular dynamics simulations and protein interactions, revisiting foundational concepts is both refreshing and essential. Recent explorations into protein-protein docking highlighted the need for a deeper understanding of amino acids, the fundamental building blocks of proteins. As I embark on this journey back to the basics, it's time to dissect each amino acid's properties and how they might influence future explorations. It's kind of daunting; after all, is there ever an endpoint in learning? But that's precisely why we dive into the complexities of amino acids: to build a robust understanding that lays the groundwork for advanced studies in protein behavior.Learning Objectives in Amino Acid Chemistry
The goal here is multifaceted. First, I’ll take a closer look at individual amino acids, particularly their distinctive side chains. Next, I want to integrate rudimentary mathematics often associated with protein structures—think Rodrigues rotation and Lennard-Jones potential. These concepts will not only reinforce the understanding of how amino acids interact but also clarify some complex mathematical occurrences in molecular simulations. If you're also delving into this field, you may find it beneficial to spotlight non-polar amino acids, reflect on the Rodrigues Rotation formula, and grasp the Lennard-Jones potential energy interactions that crucially influence how proteins fold and function.Amino Acids: The Building Blocks of Proteins
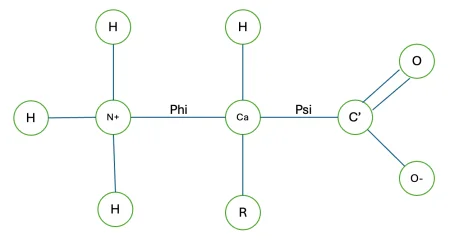
At the heart of protein structure are amino acids, characterized by a shared backbone that consists of a central α-carbon. This carbon is bonded to an amino group (–NH₂), a carboxyl group (–COOH), a hydrogen atom, and a variable side chain (R group), which ultimately defines the unique chemical identity and role of each amino acid.
Non-Polar Amino Acids: Implications for Protein Structure
Non-polar amino acids exhibit hydrophobic side chains that exhibit a tendency to steer clear of water, often clustering together in the interior of folded proteins. This behavior is vital as it contributes to the formation of a hydrophobic core that enhances protein stability. The physical properties of these amino acids—like their size and shape—play a pivotal role in how they influence backbone flexibility and how substitutions within their structure can affect active sites in enzymes. Claude has compiled an impressive table of these non-polar amino acids, including Glycine, Alanine, and Valine, among others. Each entry includes not just basic characteristics but also insights into their significance in protein function and molecular dynamics simulations. For a visual representation, here’s what we know about them:library(tibble) library(kableExtra) aa_nonpolar <- tribble( ~aa, ~aa3, ~name, ~functional_group, ~smiles_sidechain, ~charge_ph7, ~mw_da, ~pka, ~md_note, ~main_function, "G", "Gly", "Glycine", "H (none)", "[H]", "Neutral", 75.03, NA_real_, "Minimal VDW radius; unrestricted phi/psi; near-zero excluded volume", "Conformational flexibility; tight turns; active site geometry", "A", "Ala", "Alanine", "Methyl", "C", "Neutral", 89.09, NA_real_, "Low steric perturbation; high alpha-helix propensity in force fields", "Helix former; hydrophobic core; alanine-scanning mutagenesis", "V", "Val", "Valine", "Isopropyl", "CC(C)", "Neutral", 117.15, NA_real_, "Beta-branching restricts psi; favors extended beta-sheet; large gamma-carbons", "Beta-sheet core; hydrophobic packing; sickle-cell HbS Glu6Val", "L", "Leu", "Leucine", "Isobutyl", "CCC(C)C", "Neutral", 131.17, NA_real_, "Flexible chi2; common rotamers at -65/-65 and -65/175; high hydrophobic SASA", "Hydrophobic core; leucine zippers; most abundant non-polar in proteomes", "I", "Ile", "Isoleucine", "sec-Butyl", "CCC(C)", "Neutral", 131.17, NA_real_, "Beta-branching + gamma-branch; most restricted chi1/chi2; large buried SASA", "Hydrophobic core; beta-barrel interiors; transmembrane helices", "P", "Pro", "Proline", "Pyrrolidine ring", "C1CCNC1", "Neutral", 115.13, NA_real_, "Fixed phi ~-60; no backbone NH donor; cis/trans isomerism at Xaa-Pro bond", "Helix breaker; beta-turns; collagen Gly-Pro-X repeats", "F", "Phe", "Phenylalanine", "Benzyl", "Cc1ccccc1", "Neutral", 165.19, NA_real_, "Rigid aromatic ring; pi-pi stacking and cation-pi in MD energy decomposition", "Hydrophobic core; aromatic clusters; ligand binding pockets", "W", "Trp", "Tryptophan", "Indolylmethyl", "Cc1c[nH]c2ccccc12", "Neutral", 204.23, NA_real_, "Indole NH can H-bond; amphipathic at membrane interface; strong 280nm absorbance", "Membrane anchoring; fluorescence probe; ligand binding; rarest standard AA", "M", "Met", "Methionine", "Thioether", "CCSC", "Neutral", 149.20, NA_real_, "Flexible sulfur geometry; oxidizable to sulfoxide in long MD runs; check reactive FF", "Translation initiation; hydrophobic core; redox sensing" ) aa_nonpolar |> dplyr::select(aa:mw_da) |> kbl()
|
aa |
aa3 |
name |
functional_group |
smiles_sidechain |
charge_ph7 |
mw_da |
|---|---|---|---|---|---|---|
|
G |
Gly |
Glycine |
H (none) |
[H] |
Neutral |
75.03 |


